Cancel Preloader
Quality Driven
By Passion
Our Solutions
Company
Our Products
Resources
Global Reach
Industry
Submit Query
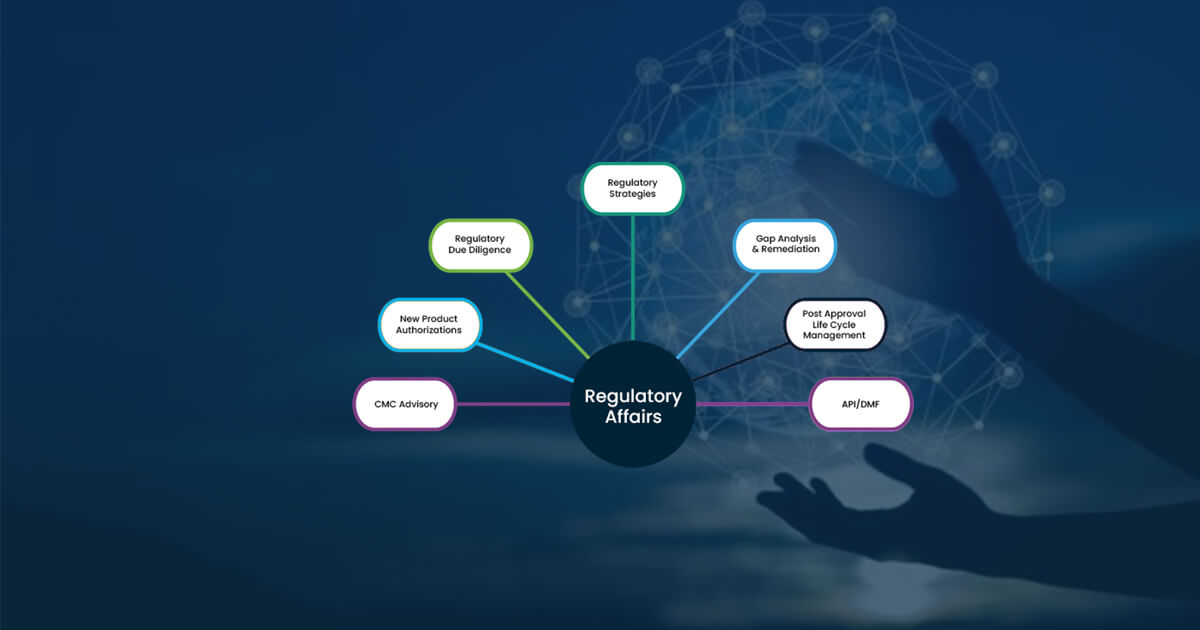
Role of Regulatory Affairs in the Pharmaceutical Industry
July 20, 2022